
Это интересно
- ОКД
- ЗКС
- ИПО
- КНПВ
- Мондиоринг
- Большой ринг
- Французский ринг
- Аджилити
- Фризби
Опрос
Полезные ссылки
РКФ

Все о дрессировке собак

Стрижка собак в Коломне

Поиск по сайту
Региональная общественная организация "Общество специалистов по нервно-мышечным болезням". Нервно мышечные заболевания журнал
Нервно-мышечные болезни, генетическое заболевание мышц: симптому, лечение
Нервно-мышечные заболевания характеризуются нарушением функции произвольной мускулатуры, утраты или снижения двигательного контроля, которое может наступать в результате поражения как собственно мышц, так и иметь вторичный характер – вследствие дисфункции нервно-мышечного соединения, поражения периферических нервов или мотонейронов спинного мозга. В клинической картине некоторых нервно-мышечных заболеваний могут присутствовать признаки поражения двигательных ядер ствола головного мозга. Поражения других участков нервной системы, приводящих к нарушению двигательного контроля, в частности пирамидного тракта, согласно общепринятому определению не относятся к нервно-мышечным заболеваниям.

Наиболее частыми симптомами нервно-мышечных заболеваний являются слабость, снижение мышечного объема (атрофия), непроизвольные мышечные подергивания, спазмы, онемение, покалывание и др. Нарушение функции нервно-мышечного соединения может вызывать опущение век (птоз), двоение в глазах (диплопия), и другие признаки мышечной слабости, которые усиливаются в течение дня. При некоторых заболеваниях могут нарушаться глотание, и даже дыхание.
Заболевания мышц: симптомы
- Прогрессирующие мышечные дистрофии – генетическое наследственные заболевание мышц, симптомы которой обычно проявляются в младенческом или детском возрасте, реже – у взрослых. Постепенно нарастает мышечная слабость, особенно заметная в произвольной мускулатуре. В эту группу входят мышечная дистрофия Бекера, врожденная мышечная дистрофия, дистальная мышечная дистрофия, мышечная дистрофия Дюшенна (наиболее часта форма миодистрофии у детей), миодистрофия Эмери-Дрейфуса, плече - лопаточная миодистрофия, миотоническая мышечная дистрофия (наиболее частая форма миодистрофии у взрослых), окулофарингеальная миодистрофия.
- Воспалительные миопатии – называют также миозитами, в основе лежит воспалительный процесс, которые приводит к слабости мышц, в их развитии подчеркивается роль аутоиммунных нарушений, иногда сочетаются с другими аутоиммунными заболеваниями. К ним относятся дерматомиозит, полимиозит, миозит с включениями.
- Митохондриальные миопатии – возникают в результате структурных или биохимических дефектов митохондрий. Синдром Кернса-Сайре, миоклонус эпилепсия с «разорванными красными волокнами», митохондриальная энцефаломиопатия.
- Миотонии - врожденная миотония или болезнь Томсена, дистрофическая миотония, врожденная парамиотония, нейромиотония или болезнь Исаакса
- Другие миопатии – болезнь центрального стержня, миотубудярная миопатия, немалиновая миопатия, периодический гиперкалиемический и гипокалиемический паралич, эндокринные миопатии
Заболевания нервно-мышечного соединения
Вызывают дисфункцию нормальной синаптической передачи импульсов с нервных окончаний на мышечные волокна. В основе заболевания может лежать аутоиммунный процесс.
- миастения гравис
- синдром Ламберта-Итона
- врожденный миастенический синдром
Заболевания периферических нервов
- Мононейропатии - поражение одного нерва, наиболее частой причиной является компрессионное воздействие (туннельные синдромы), травматические повреждения
- Множественные мононейропатии - мультифокальные поражения периферических нервов, связаны обычно с системными или инфекционными заболеваниями, паранеопластическими синдромами
- Полинейропатии - диффузные, симметричные поражения периферических нервов, обычно преобладаюшие в дистальных отделах, почти всегда в клинической картине присутствуют расстройства чувствительности . Могут быть острыми (синдром Гиейна-Барре), хроническими (хроническая воспалительная демиелинизируюшая полинейропатия), приобретенными (токсические, диабетические, паранеопластические) или наследственными (перонеальная мышечная атрофия или болезнь Шарко-Мари-Тус, болезнь Дежерина-Сотта, атаксия Фридрейха).
- Плексопатии - поражения сплетений верхних и нижних конечностей (плечевого и пояснично-крестцового), наиболее частой причиной которых являются травматическое или компрессионное воздействие
- Радикулопатии и полирадикулопатии - поражения двигательных или чувствительных спинальных корешков
Заболевания двигательного нейрона
Прогрессирующее дегенеративное поражение мотонейронов, которое наиболее заметно приводит к нарушению двигательного контроля верхних или нижних конечностей, а также бульбарным расстройствам. Чаще начинаются в среднем возрасте, симптомы могут включать слабость в конечностях, нарушение глотания, речи, походки, слабость лицевой мускулатуры, мышечные спазмы. В эту группу входят, в частности:
- боковой амиотрофический склероз (БАС)
- спинальная мышечная атрофия взрослых
- спинальная мышечная атрофия у младенцев или болезнь Верднига-Гоффмана
- юношеская спинальная мышечная атрофия или болезнь Кугельберга-Веландера
- бульбоспинальная мышечная атрофия или болезнь Кеннеди
Диагноз ставится на основе истории заболевания, тщательного неврологического осмотра, в большинстве случаев используется электромиографическое (ЭМГ) исследование, при сочетании с поражением центрального мотонейрона или для исключения его может применяться транскраниальная магнитная стимуляция, при подозрении на наследственные формы проводится анализ ДНК, аутоимунный характер процесса требует определения специфических антител, может проводиться биопсия участка мышцы, при первично-мышечных поражениях проверяется уровень содержания креатинфосфокиназы (КФК), в последнее время набирает также популярность ультразвуковое исследования мышц и периферических нервов. Диагностический алгоритм, выбор дополнительных исследований зависят от особенностей клинического паттерна и локализации поражения - мышца, нерв, сплетения, корешки, двигательные нейроны.
www.nevromed.ru
Неврология
 Наследственные миотонические синдромы (НМС) - группа генетически гетерогенных заболеваний ионных каналов хлора и натрия (каналопатии), характеризующиеся повышенной возбудимостью мембраны мышечных волокон, проявляющиеся миотоническими феноменами с постоянной или транзиторной слабостью скелетной мускулатуры. В современной классификации НМС представлены различными генетическими формами [1] дистрофических (ДМ) и [2] недистрофических миотоний (НДМ).
Наследственные миотонические синдромы (НМС) - группа генетически гетерогенных заболеваний ионных каналов хлора и натрия (каналопатии), характеризующиеся повышенной возбудимостью мембраны мышечных волокон, проявляющиеся миотоническими феноменами с постоянной или транзиторной слабостью скелетной мускулатуры. В современной классификации НМС представлены различными генетическими формами [1] дистрофических (ДМ) и [2] недистрофических миотоний (НДМ).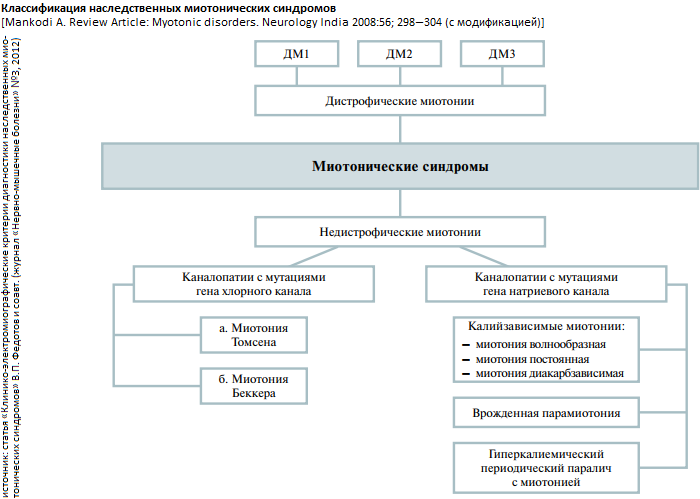

 о клинической и ЭМГ феноменологии миотонии вы можете прочитать в статье «Клинико-диагностические критерии миотонии» Н. А. Шнайдер; ГБОУ ВПО Красноярский государственный медицинский университет имени проф. В. Ф. Войно-Ясенецкого Министерства здравоохранения РФ, кафедра медицинской генетики и клинической нейрофизиологии ИПО (журнал «Сибирское медицинское обозрение» №3, 2016) [читать]
о клинической и ЭМГ феноменологии миотонии вы можете прочитать в статье «Клинико-диагностические критерии миотонии» Н. А. Шнайдер; ГБОУ ВПО Красноярский государственный медицинский университет имени проф. В. Ф. Войно-Ясенецкого Министерства здравоохранения РФ, кафедра медицинской генетики и клинической нейрофизиологии ИПО (журнал «Сибирское медицинское обозрение» №3, 2016) [читать]Недистрофические миотонии. Самая распространенная форма НДМ - врожденная миотония (ВМ), с распространенностью от 1 до 9,4 на 100 тыс. населения, что зависит от страны и этнической принадлежности. Происхождение ВМ обусловлено мутациями в гене хлорного канала CLCN1 (локус 7q35) в поперечно-полосатых мышцах (нарушается проводимость ионов Cl- в клетку, что ведет к повышенной возбудимости мышечной мембраны и мышечной ригидности). ВМ клинически проявляются:
 [1] генерализованными миотоническими феноменами; [2] гипертрофией скелетной мускулатуры; [3] транзиторной слабостью; [4] дебютом в раннем возрасте; [5] стационарным течением и благоприятным прогнозом.
[1] генерализованными миотоническими феноменами; [2] гипертрофией скелетной мускулатуры; [3] транзиторной слабостью; [4] дебютом в раннем возрасте; [5] стационарным течением и благоприятным прогнозом.
На сегодня выделяют 2 формы ВМ. Первой описана миотония Томсена (МТ) с аутосомно-доминантным типом наследования и лишь 100 лет спустя - миотония Беккера (МБ) с аутосомно-рецессивным типом наследования. Молекулярно-генетические данные показали, что ранее считавшаяся редкой ВМ Беккера встречается даже чаще, чем ВМ Томсена. При дебюте в раннем возрасте пациенты с МТ и МБ имеют, как было указано выше, гипертрофию скелетных мышц («атлетическое сложение», более характерное для ВМ Томсена), иногда легкую миалгию, слабость, задержку мышечного расслабления, а также относительно стационарное течение и благоприятный прогноз. То есть больные с МТ и МБ имеют однотипные клинические проявления, различаясь лишь степенью их выраженности (ВМ Беккера считают несколько более тяжелой, но не в каждом случае).
Дистрофические миотонии (или ДМ, или сongenital myotonic dystrophy, myotonic dystrophy - DM) является мультисистемным заболеванием, при котором мутация затрагивает развитие и функционирование различных органов и тканей: гладкой и скелетной мышечной ткани, сердца, органа зрения (глаза), головного мозга. Это наиболее распространенное заболевание из класса миотоний. Клиническая картина дистрофической миотонии складывается из 3 синдромов: [1] миотонический синдром;[2] дистрофический синдром; [3] синдром вегетативно-трофических нарушений.
Таким образом, ключевая особенность ДМ - сочетание миотонии, которая характеризуется отсроченным расслаблением после мышечного сокращения, и прогрессирующей мышечной слабости, дистрофии (атрофии). МД характеризуется варьиру ющим началом заболевания: от пренатального периода до 50 - 60 лет. Различают четыре формы по возрастному «пику» начала заболевания: [1] врожденная (син.: конгенитальная ДМ или congenital myotonic dystrophy [CmyD]; клиническая симптоматика развивается сразу после рождения), [2] юношеская (син.: ювенильная МД или juvenile DM; с дебютом от 1 года до подросткового возраста), [3] классическая (син.: ДМ взрослых или adult DM; - с дебютом у индивидуумов старше 20, но моложе 40 лет) и [4] минимальная (син.: МД с поздним дебютом или DM with late onset; у индивидуумов старше 40 лет [как правило, до 60 лет] и с более легким течением). Это объясняется различиями в числе тринуклеотидных повто ров (CTG) в локусе гена МД, кодирующего синтез миотонин-протеинкиназы. Кроме различия в возрасте начала проявления болезни имеются достаточно различные клинические признаки разных подтипов этого заболевания.
До 1994 года ДМ считалась однородным заболеванием. Однако в последние годы после идентификации различных мутаций при сходной клинической симптоматике, напоминающей дистрофическую миотонию, было показано, что это гетерогенное заболевание, представленное тремя подтипами: DM1 (мутация 19q13.3), DM2 (мутация 3q21) и DM3 (мутация 15q21-q24). Существуют отдельные исследования, подтверждающие наличие четвертого подтипа DM и др. (DM4, DMX). Наиболее часто встречается (среди всех миотоний, как среди ДМ, так и среди НДМ) DM1 - ДМ Россолимо-Куршмана-Штейнерта-Баттена (с частотой 13,5 на 100000 живых новорожденных; распространенность в больших популяциях около 1 : 8000). Распространенность DM2 (болезнь Thornton-Griggs-Moxley) и DM3 в настоящее время недостаточно изучена.

 читайте также пост: Креатинкиназа (справочник невролога) (на laesus-de-liro.livejournal.com) [читать]В отличие от большинства других наследственных нервно-мышечных заболеваний (и от других форм миотоний, в частности) клинические проявления ДМ вариабельны и могут различаться от человека к человеку даже в пределах одной семьи. Больные с ДМ могут сталкиваться с большим числом разнообразных (необычных для миотонии) проблем, к которым можно отнести низкий уровень жизненной активности, пассивность, депрессию, облысение, нарушения со стороны желудочно-кишечного тракта, сексуальные проблемы, что в ряде случаев приводит к возникновению недопонимания и даже к конфликтным ситуациям между пациентом и лечащим врачом. Следует помнить, что пассивное поведение пациента является по большей мере реальным проявлением данного заболевания, а не желанием человека.
читайте также пост: Креатинкиназа (справочник невролога) (на laesus-de-liro.livejournal.com) [читать]В отличие от большинства других наследственных нервно-мышечных заболеваний (и от других форм миотоний, в частности) клинические проявления ДМ вариабельны и могут различаться от человека к человеку даже в пределах одной семьи. Больные с ДМ могут сталкиваться с большим числом разнообразных (необычных для миотонии) проблем, к которым можно отнести низкий уровень жизненной активности, пассивность, депрессию, облысение, нарушения со стороны желудочно-кишечного тракта, сексуальные проблемы, что в ряде случаев приводит к возникновению недопонимания и даже к конфликтным ситуациям между пациентом и лечащим врачом. Следует помнить, что пассивное поведение пациента является по большей мере реальным проявлением данного заболевания, а не желанием человека.подробнее о DM1, DM2 и DM3 читайте [1] в статье «Клинико-генетическая гетерогенность дистрофической миотонии» Н.А. Шнайдер, Е.А. Козулина, Д.В. Дмитренко; Кафедра медицинской генетики и клинической нейрофизиологии Института последипломного образования ГОУ ВПО «Красноярская гос. мед. академия Федерального агентства по здраво-охранению и социальному развитию», Россия (Международный неврологический журнал, №3, 2007) [читать] (или [читать]) и [2] в статье «Миотоническая дистрофия. Современное представление и собственное наблюдение» Т.И. Стеценко, Национальная медицинская академия последипломного образования имени П.Л. Шупика, г. Киев, Украина (журнал «Современ-ная педиатрия» №1, 2014) [читать]
 Обратите внимание! Для НДМ не характерно развитие парезов конечностей и мышечных атрофий в отличие от больных с ДМ1, у которых вялые дистальные парезы рук и ног служат главной причиной инвалидизации. Тем не менее, у больных с НДМ (форма Томсена, Беккера) специфическими симптомами могут выступать исходная неловкость и мышечная слабость в кистях, обусловленная типичными миотоническими задержками и исчезающая после нескольких повторных произвольных сокращений мышц. Симптом получил наименование «транзиторная слабость», а уменьшение выраженности миотонии при повторных мышечных сокращениях - «феномен врабатывания». Транзиторная слабость отсутствует у больных с ДМ1, а слабость в мышцах кисти остается неизменной даже после уменьшения миотонических проявлений. Наличие транзиторной слабости у больных с НДМ связывали с дебютом ДМ1, что затрудняло дифференциальную диагностику, приводило к неверным нозологическим трактовкам и неадекватным генетическим прогнозам в семьях пробандов. Все это послужило поводом к необходимости выработки объективных критериев наличия или отсутствия транзиторной слабости. Проведение теста с ритмической стимуляцией (РС) с частотой 10 - 60 Гц у больных с НМС выявило декремент М-ответа с восстановлением его амплитуды после тетанизации. Кроме того, при проведении РС у больных с НМС на примере мышц кисти, иннервируемых локтевым нервом, выявляется зависимость между величиной декремента амплитуды М-ответов и выраженностью транзиторной слабости. Выявленный декремент М-ответа при РС в генотипированной группе больных с НДМ объясняется нарушением функции хлорных каналов. [!!!] Таким образом, декремент амплитуды М-ответов может быть информативным показателем в алгоритме диагностического поиска мутаций в гене хлорного канала у пациентов с НМС.
Обратите внимание! Для НДМ не характерно развитие парезов конечностей и мышечных атрофий в отличие от больных с ДМ1, у которых вялые дистальные парезы рук и ног служат главной причиной инвалидизации. Тем не менее, у больных с НДМ (форма Томсена, Беккера) специфическими симптомами могут выступать исходная неловкость и мышечная слабость в кистях, обусловленная типичными миотоническими задержками и исчезающая после нескольких повторных произвольных сокращений мышц. Симптом получил наименование «транзиторная слабость», а уменьшение выраженности миотонии при повторных мышечных сокращениях - «феномен врабатывания». Транзиторная слабость отсутствует у больных с ДМ1, а слабость в мышцах кисти остается неизменной даже после уменьшения миотонических проявлений. Наличие транзиторной слабости у больных с НДМ связывали с дебютом ДМ1, что затрудняло дифференциальную диагностику, приводило к неверным нозологическим трактовкам и неадекватным генетическим прогнозам в семьях пробандов. Все это послужило поводом к необходимости выработки объективных критериев наличия или отсутствия транзиторной слабости. Проведение теста с ритмической стимуляцией (РС) с частотой 10 - 60 Гц у больных с НМС выявило декремент М-ответа с восстановлением его амплитуды после тетанизации. Кроме того, при проведении РС у больных с НМС на примере мышц кисти, иннервируемых локтевым нервом, выявляется зависимость между величиной декремента амплитуды М-ответов и выраженностью транзиторной слабости. Выявленный декремент М-ответа при РС в генотипированной группе больных с НДМ объясняется нарушением функции хлорных каналов. [!!!] Таким образом, декремент амплитуды М-ответов может быть информативным показателем в алгоритме диагностического поиска мутаций в гене хлорного канала у пациентов с НМС.


статья «Клинико-электромиографические критерии диагностики наследственных миотонических синдромов» В.П. Федотов, С.А. Курбатов, Е.А. Иванова, Н.М. Галеева, А.В. Поляков; Воронежская медико-генетическая консультация, БУЗ ВО «Воронежская областная клиническая больница №1»; ФГБУ «Медико-генетический научный центр» РАМН, Москва (журнал «Нервно-мышеч-ные болезни» №3, 2012) [читать];
статья «Случай миотонии Беккера с псевдодоминантным типом наследования: современные подходы к дифференциальной диагностике миотоний Томсена и Беккера» С.А. Курбатов, С.C. Никитин, С.Н. Иллариошкин, П. Гундорова, А.В. Поляков; АУЗ ВО «Воронежский областной клинический консультативно-диагностический центр»; Региональная общественная организация Общество специалистов по нервно-мышечным болезням», Медицинский центр «Практическая неврология», Москва; ФГБНУ «Научный центр неврологии», Москва; ФГБНУ «Медико-генетический научный центр», Москва (журнал «Нервно-мышечные болезни» №1, 2016) [читать];
статья «Случай миотонии Беккера с эквинной деформацией стоп» Г.Е. Руденская, Е.А. Иванова, Н.М. Галеева; Медико-генети-ческий научный центр РАМН, Москва (Журнал неврологии и психиатрии, №8, 2012) [читать];
статья «Миотонии» (сокращенное изложение) Teruyuki Kurihara, Internal Medicine 2005; 44:1027-1032 (подготовил Ю. Матвиенко) [читать];
диссертация на соискание ученой степени кандидата биологических наук «Генетическая вариабельность локуса миотонин-протеинкиназы в якутской популяции» Степанова С.К., ФГБНУ «Якутский научный центр комплексных медицинских проблем» ФГБНУ «Научно-исследовательский институт медицинской генетики»; Томск, 2015 [читать];
статья «Миотоническая дистрофия 2-го типа» Г.Е. Руденская, А.В. Поляков; ФГБУ «Медико-генетический научный центр» РАМН, Москва (журнал «Анналы клинической и экспериментальной неврологии» №2, 2012) [читать];
статья «Дистрофическая миотония Россолимо-Штейнерта-Куршмана» Авдей Г.М., Кулеш С.Д., Шумскас М.С., Авдей С.А.; УО «Гродненский государственный медицинский университет»; УЗ «Гродненская областная клиническая больница», г. Гродно (Уральский медицинский журнал, №9, 2014) [читать];
статья «Случай дистрофической миотонии 1-го типа с утяжелением клиники по линии отца» Курбатов С.А., Федотов В.П., Галеева Н.М., Забненкова В.В., Поляков А.В.; АУЗ ВО «Воронежский областной клинический консультативно-диагностический центр»; БУЗ ВО «Воронежская областная клиническая больница № 1»; ФГБНУ «Медико-генетический научный центр», Москва (журнал «Анналы клинической и экспериментальной неврологии» №2, 2015) [читать];
статья «Случай поздней диагностики дистрофической миотонии Россолимо-Штейнерта-Куршмана» Ю.Н. Быков, Ю.Н. Васильев, О.П. Панасюк, А.Ч. Янгутова; Иркутский государственный медицинский университет, кафедра нервных болезней (Сибирский медицинский журнал, №2, 2016) [читать];
статья «Клинико-генетическая гетерогенность хондродистрофической миотонии» Н.А. Шнайдер, Красноярский ГМУ им. проф. В.Ф. Войно-Ясенецкого, Красноярск (журнал «Нервно-мышечные болезни» №2, 2012) [читать];
статья «Профилактика аспирационной пневмонии у больных дистрофической миотонией с орофарингеальной дисфагией» Е.А. Бахтина, Н.А. Шнайдер, Т.Л. Камоза, Е.А. Козулина; Красноярская государственная медицинская академия им. В.Ф. Войно-Ясенецкого, кафедра медицинской генетики и клинической нейрофизиологии Института последипломного образования; Красноярский государственный торгово-экономический институт, кафедра технологии питания (журнал «Сибирское медицинское обозрение» №3, 2008) [читать];
статья «Мультидисциплинарный подход к ведению беременных, больных дистрофической миотонией» Н.А. Шнайдер, Е.А. Козулина, В.А. Шульман, С.Ю. Никулина, Р.А. Бутьянов, Е.А. Бахтина; Красноярская государственная медицинская академия им. В.Ф. Войно-Ясенецкого Росздрава; кафедра медицинской генетики и клинической нейрофизиологии ИПО; кафедра внутренних болезней №1 (журнал «Сибирское медицинское обозрение» № 4, 2008) [читать]
laesus-de-liro.livejournal.com
| 16 - 19 мая 2018 | Школа миологии-2018. Наследственные болезни периферического нейромоторного аппарата в ежедневной практике | Россия, Москва |
| 22 - 23 декабря 2017 | Нередкие редкие болезни: болезни периферических нервов | Россия, Москва |
| 28 - 29 октября 2016 | Научно-практическая конференция с международным участием «Аутоиммунные поражения периферических нервов и мышц» | Россия, Москва |
| 04 - 05 июня 2016 | Обучение по теме: «Современные методы диагностики и лечения компрессионных синдромов плечевого сплетения» | Россия, город Москва, Москва |
| 29 - 30 апреля 2016 | Научно-практическая конференция с международным участием «Поясно-конечностные мышечные дистрофии: от осмотра до диагноза» | Россия, Москва |
| 23 - 24 октября 2015 | Научно-практическая конференция с международным участием «Новые технологии в диагностике и лечении болезней нервно-мышечной системы» 23-24 октября | Россия, Москва |
| 04 - 05 июня 2015 | Первая Московская конференция с международным участием «ФУНДАМЕНТАЛЬНАЯ И КЛИНИЧЕСКАЯ НЕВРОЛОГИЯ. ТРАНСКРАНИАЛЬНАЯ МАГНИТНАЯ СТИМУЛЯЦИЯ: ДОСТИЖЕНИЯ И ПЕРСПЕКТИВЫ» | Москва |
| 01 июня 2015 | I Национальная научно-практическая конференция по нарушениям глотания (с международным участием) «ПРОБЛЕМЫ ВЫЯВЛЕНИЯ, ДИАГНОСТИКИ И ВЕДЕНИЯ ПАЦИЕНТОВ С НАРУШЕНИЯМИ ГЛОТАНИЯ» | Россия, город Москва, Москва |
| 14 марта 2015 | Cеминар "Генетика эпилепсии" | Москва |
| 14 ноября 2014 | Конференция с международным участием «Инновации в эпилептологии 5+» | Москва |
| 24 - 26 апреля 2014 | Научно-практическая конференция "Нейрофизиологические исследования в клинике" | Москва |
| 28 - 29 марта 2014 | Российская научно-практическая конференция с международным участием и специализированная выставка «Дифференциальный диагноз в клинике нервно-мышечных болезней» | Москва |
| 23 декабря 2013 | Cеминар «Нейропатия лицевого нерва» | г. Москва |
| 08 ноября 2013 | Российская Конференция с международным участием «Объединения врачей-эпилептологов и пациентов» «Инновации в эпилептологии IV+» | Москва |
| 13 - 17 мая 2013 | Авторский курс повышения квалификации: "Клиническая электронейромиография в диагностике". | Москва |
| 26 апреля 2013 | Научно-практическая конференция "Алгоритмы диагностики наследственных нервно-мышечных болезней" | Москва, Россия |
| 11 - 12 апреля 2013 | Научно-практическая конференция "Нейрофизиологические исследования в клинике" | Москва |
| 16 марта 2013 | Конференция «Внутривенная иммунотерапия в неврологии и ревматологии: теоретические аспекты, отечественный опыт применения» | Ярославль |
| 25 января 2013 | XIV Научно-практическая нейроофтальмологическая конференция "Актуальные вопросы нейроофтальмологии" | Москва |
| 14 декабря 2012 | 1-ое заседание Российского Экспертного Совета по электромиографии | Москва |
| 08 - 09 июня 2012 | Северо-Западная Конференция с международным участием "Объединения врачей-эпилептологов и пациентов" "Содружество врача и пациента, страдающего эпилепсией" | Санкт-Петербург, Россия |
| 25 - 26 мая 2012 | I Московский Международный Симпозиум по нейрореанимации | Москва, Россия |
| 29 - 30 августа 2011 | Курс лекций и клинических разборов Anthony Amato (Бостон) | Москва |
www.neuromuscular.ru
Нервно-мышечные заболевания
Заболевания периферических нервов
Нейромышечный синапс
Миопатии
Клинические синдромы
Заболевания периферических нервов
Определения
Мононейропатия. Изолированное поражение периферических нервов, например, при сдавлении, травме, нарушении кровоснабжения (поражение vasa vasorum).
Системные заболевания, поражающие нервы, чувствительные к сдавлению, например сахарный диабет, или патологические состояния, вызывающие диффузные нарушения сосудистого русла (васкулиты), способны вызывать мультифокальную нейропатию (или множественную полинейронатию).
Полинейропатия. Одновременное множественные поражение периферических нервов вследствие воспалительных процессов, метаболических нарушений или токсических воздействий. Клинически проявляется диффузным, симметричным поражением периферических нервов. В первую очередь страдают дистальные отделы конечностей, причем нижние конечности поражаются раньше верхних.
Мононейропатии
Наиболее часто встречаются следующие мононейропатии.
Синдром запястного канала
Компрессия срединного нерва в области запястья при его прохождении через канал может произойти:
- изолированно; например, у пациентов с избыточными физическим нагрузками (связанными с характером трудовой деятельности)
- при заболеваниях, характеризующихся повышенной чувствительностью нервных стволов к внешнему воздействию (сдавлению)
- при сдавлении нервного ствола в области запястного канала гипертрофированными тканями (табл. 1).
Таблица 1. Состояния, ассоциированные с синдромом запястного канала
|
Беременность Сахарный диабет Локальные деформации вследствие остеоартрита, переломов костей Рематоидный артрит Микседема Акромегалия Амилоидоз |
Клинические проявления синдрома запястного капала:
- боль в кисти или предплечье, особенно ночью или при напряжении
- парез (паралич) и гипотрофия мышц возвышения большого пальца (thenar)
- снижение чувствительности в зоне иннервации срединного нерва (рис. 1)
- парестезии по ходу срединного нерва, которые возникают при постукивании в области запястного канала (симптом Тинеля)
- как правило, двустороннее поражение.

Рис. 1. Распределение зон иннервации срединного, локтевого и лучевого нервов на поверхности плеча и предплечья
Диагноз может быть подтвержден при помощи электрофизиологических исследований. Определение содержания в крови глюкозы, гормонов щитовидной железы, СОЭ, могут помочь в установлении правильного диагноза.
Лечение определяется тяжестью состояния больного. Основные лечебные мероприятия:
- фиксация мышц, особенно ночью, в частично вытянутом состоянии, кисть должна при этом находиться в состоянии разгибания
- мочегонные средства — эффект неясен
- введение кортикостероидов в просвет запястного канала
- хирургическая декомпрессия срединного нерва.
Нейропатия локтевого нерва
Локтевой нерв может быть подвержен сдавлению на различном уровне, однако наиболее часто это случается в области локтевого сустава.
Клинические проявления:
- боли и/или парестезии (покалывающего характера), распространяющиеся от локтя вниз по локтевой поверхности к предплечью
- паралич или слабость внутренних мышц кисти (поражение мышц возвышения большого пальца)
- снижение чувствительности в зоне иннервации локтевого нерва (рис. 1)
- при хроническом поражении формируется когтистая кисть.
Определение скорости проведения импульса при помощи электронейрографического исследования позволяет точно установить локализацию поражения локтевого нерва.
При нетяжелом поражении может быть эффективна фиксация руки на ночь, выпрямленной в локтевом суставе, что обеспечивает уменьшение сдавления нервного ствола. При более тяжелом поражении положительный результат обеспечивает хирургическая декомпрессия или транспозиция локтевого нерва, однако полный регресс неврологической симптоматики наблюдается не всегда. Оперативное вмешательство показано при постоянной травматизации локтевого нерва, которая сопровождается постоянным болевым синдромом и/или прогрессирующими двигательными нарушениями (парез).
Парез лучевого нерва
Сдавление лучевого нерва в верхней части предплечья может привести к острому развитию синдрома «свисающей кисти», при этом иногда наблюдается утрата чувствительности в зоне иннервации лучевого нерва (рис. 1). Как правило, данное поражение является следствием длительного пребывания предплечья в непривычном положении, например при свисающей в неудобном положении руке с поручня кресла при алкогольном опьянении («паралич субботнего вечера»).
Парез плечевого сплетения
Помимо острой травмы плечевого сплетения (например, в результате родовой травмы или дорожно-транспортного происшествия у мотоциклистов) поражение плечевого сплетения может быть обусловлено другими причинами. Поражение верхнего отдела сплетения носит название паралича Эрба, а нижнего - паралича Клюмпке.
Добавочное ребро
Добавочное ребро или гипертрофированная соединительная ткань может быть причиной компрессии плечевого сплетения в области верхней апертуры грудной клетки. На определенном этапе развития неврологии и нейрохирургии имела место гипердиагностика данного состояния и, как следствие, высокая частота необоснованных хирургических вмешательств. На сегодняшний день считается, что оперативное вмешательство показано пациентам с нарастающим парезом внутренних мышц предплечья, выраженной утратой чувствительности (по ходу локтевого нерва) и с диагнозом, подтвержденным электрофизиологическими методами обследования. Визуализация плечевого сплетения при помощи МРТ обычно неэффективна. При рентгенографическом обследовании возможно выявление добавочного ребра, но сдавление нервного ствола фиброзной тканью визуализировать не удается.
Опухоль Пенкоста
Бронхогенная карцинома верхушки легкого может прорастать в нижние корешки плечевого сплетения, вызывая усиливающуюся боль в одноименной руке, дистальный паралич и гипотрофию, а также снижение чувствительности в дерматомах С7, С8 и Th20. Возможен также синдром Горнера вследствие поражения преганглионарных вегетативных волокон. Симптоматика имеет сходство с первичными и метастатическими опухолями.
Диагностические трудности возникают при поражении сплетения у больных с карциномой молочной железы после проведенного курса лучевой терапии, так как неврологический дефицит может быть следствием распространения опухоли или радиационной плексопатии.
Идиопатическая плечевая плексопатия (невралгическая амиотрофия или нейропатия плечевого нерва)
Состояние характеризуется острой болью в плече и предплечье. Очевидных причин этому нет, хотя заболевание может возникнуть после прививки или операции. После регресса болей (через несколько дней или недель) появляется частичный паралич и слабость окололопаточной группы мышц, а также более удаленных мышечных групп верхней конечности. Особенно подвержена поражению передняя лестничная мышца, атрофия которой сопровождается развитием крыловидных лопаток (рис. 2). Поражение, как правило, одностороннее, с минимальными чувствительными нарушениями. Электрофизиологические исследования зачастую малоэффективны, хотя могут выявляться признаки денервации пораженных мышц. Состав СМЖ не изменен. Специфического лечения не существует, у большинства больных через 1,5-2 года наступает спонтанное выздоровление.

Рис. 2. Крыловидные лопатки
Парестетическая мералгия
Компрессия латерального кожного нерва бедра, проходящего под паховой связкой; характеризуется потерей чувствительности в соответствующей области (рис. 3). Начало заболевания связано, в частности, с изменением веса пациента (увеличение или уменьшение).

Рис. 3. Парестетическая мералгия. Схема распределения нарушений чувствительности при поражении латерального кожного бедренного нерва
Латеральный подколенный паралич
Подколенный нерв подвержен компрессионным повреждениям в области, где он огибает шейку малоберцовой кости. Проявляется синдромом свисающей стопы (вследствие пареза разгибателя стопы). Одновременно появляются слабость при тыльном разгибании и отведении стопы с утратой чувствительности разной степени выраженности. Данное состояние часто встречается у иммобилизованных пациентов и у пациентов с повышенной чувствительностью нервных стволов к сдавлению, например при сахарном диабете. Свисающая стопа может быть следствием поражения поясничного корешка (обычно L5). Следует отличать данный синдром от поражения малоберцового нерва, для которого характерна сохранная внутренняя ротация стопы, так как задняя большеберцовая мышца иннервируется большеберцовым нервом, а не малоберцовым. Однако требуется электрофизиологическое исследование для уточнения локализации поражения нерва. Повреждение малоберцового нерва обычно обратимо, так как вызывается нарушением проводимости (нейрапраксия). Положительный эффект оказывает фиксация стопы лонгетой.
Мультифокальная нейропатия
Причины мультифокальной нейропатии (множественный мононеврит):
- злокачественная инфильтрация (карцинома или лимфома)
- васкулит или заболевание соединительной ткани:
- ревматоидный артрит
- системная красная волчанка
- узелковый периартрит
- гранулематоз Вегенера;
- саркоидоз
- диабет
- инфекционные заболевания:
- проказа
- опоясывающий герпес
- ВИЧ
- болезнь Лайма;
- наследственная нейропатия со склонностью к параличам от сдав-ления.
Наиболее частой причиной мультифокальной нейропатии является васкулит с болями, слабостью и гипестезией в зонах иннервации нескольких периферических нервов. Чаще поражаются нижние конечности. Поражения отдельных нервов постепенно накапливаются, проявляясь асимметричным поражение конечностей.
Полинейропатии
Диффузное поражение периферических нервов может быть разделено на группы с поражением двигательных, чувствительных или смешанных нервов. Существует патофизиологическая классификация полинейропатии, основным критерием которой является преобладание поражения миелиновой оболочки или непосредственно нервного ствола нерва (демнелинизирующая или аксональная нейропатия соответственно). Причины полинейропатии приведены в табл. 2.
Таблица 2. Причины полинейропатии
|
Наследственная предрасположенность Инфекционные заболевания Проказа Дифтерия ВИЧ Болезнь Лайма Воспалительные процессы Синдром Гийена-Барре Хроническая воспалительная демиелинизирующая полинейропатия Саркоидоз Синдром Шегрена Васкулиты (например, волчанка, полиартериит) Новообразования Паранеопластические процессы Парапротеинемические процессы Метаболические расстройства Диабет Уремия Микседема Отложения амилоида Неправильное питание Дефицит витаминов, в особенности тиамина, ниацина и витамина В12 Отравления Например, алкоголем, свинцом, мышьяком, золотом, ртутью, таллием, инсектицидами, гексаном Лекарственные препараты Например, изониазид, винкристин, цисплатин, метронидазол, нитрофураны, фенитоин, амиодарон |
У пациентов могут развиться онемение и/или парез дистальных отделов конечностей. Двигательные нарушения характеризуются вялыми парезами и мышечными атрофиями. Длительно развивающиеся нейропатии могут привести к деформации стоп и кистей рук (полая стопа — рис. 4 и когтистая кисть соответственно). Тяжелое поражение сенсорных волокон может сопровождаться развитием нейропатических язв и деформаций суставов (рис. 5). Возможны сопутствующие вегетативные расстройства. Клинические признаки такие же, как и при распространенном поражении периферических мотонейронов с вялыми параличами, гипотонией и снижением сухожильных рефлексов. Утрата проприоцептивной чувствительности в дистальных отделах конечностей может сопровождаться сенсорной атаксией. Характерно снижение болевой, температурной и тактильной чувствительности но типу «носков и перчаток». В ряде случаев можно обнаружить утолщение периферических нервов. Тактика обследования пациентов с полинейропатиями приведена в табл. 3.

Рис. 4. Полая стопа

Рис. 5. Нейропатия правой лодыжки (слева) и ступни (справа; артропатия Шарко)
Таблица 3. Обследование больного с нейропатией
|
Исследование крови Клинический анализ с подсчетом форменных элементов, СОЭ, содержание глюкозы, электролитов, мочевины, печеночных ферментов и гормонов щитовидной железы, витамина В12, электрофорез сывороточных белков, определение аутоантител Исследование мочи Микроскопический анализ для подтверждения васкулита, определение содержания глюкозы, порфиринов, белка Бена-Джонса Исследование СМЖ Повышенное содержание белка, в частности при воспалительных нейропатиях Нейрофизиологическое обследование Изучение скорости проведения по двигательным и чувствительным нервам и ЭМГ Рентгенография органов грудной клетки Для исключения саркоидоза, карциномы Специальные обследования для отдельных пациентов Биопсия периферических нервных волокон при неизвестном характере нейропатии и ухудшении состояния больного. Проводится для подтверждения наличия васкулита, проказы и хронической воспалительной демиелинизирующей полинейропатии. Биопсия костного мозга, обследование скелета при подозрении на миеломную болезнь. При определенных состояниях — специфические анализы крови, например при наследственных нейропатиях — анализ ДНК, при врожденных нарушениях метаболизма — обнаружение ферментов из лейкоцитов, при болезни Лайма — обнаружение антител к боррелии. |
Лечение больных с полинейропатиями определяется в первую очередь причинами заболевания. Пациенты с воспалительными полинейропатиями требуют госпитализации в специализированные отделения. Больному с острой воспалительной демиелинизирующей полинейропатией (синдром Гийена-Барре) может потребоваться реанимационная помощь. Хроническая воспалительная демнелинизирующая полинейропатия (ХВДП) и полиневропатия при васкулитах требуют применения кортикостероидов и/или иммуномодулирующих препаратов, включая иммуносупрессоры (азатиоприн, циклофосфамид или циклоспорин), внутривенное введение иммуноглобулина или плазмаферез. Симптоматическое лечение позволяет снизить вероятность осложнений, включая нарушения вегетативных функций и болевые синдромы.
Важно отличать синдром Гийена-Барре и ХВПД — демиелини-зирующие заболевания периферических нервов от демиелинизации ЦНС (табл. 17.4).
Таблица 4. Заболевания, приводящие к демиелинизации. Классификация в соответствии с локализацией основного очага поражения и характером течения заболевания
|
Центральная нервная система |
Периферическая нервная система | |
|
Острые |
Острый рассеянный энцефаломиелит (ОРЭМ) — редко |
Синдром Гийена-Барре — достаточно часто |
|
Хронические |
Рассеянный склероз —часто |
Хроническая воспалительная демиелинизирующая полинейропатия (ХВДП) — редко |
Нейромышечный синапс
Миастения
Аутоиммунное заболевание, при котором у большинства пациентов выявляются циркулирующие антитела к ацетилхолиновым рецепторам нейромышечных синапсов (рис. 6). В качестве причины возможна патология тимуса (гиперплазия, атрофия или опухоль — тимома). Данное заболевание встречается относительно редко, в среднем в год регистрируется 0,4 случая на 100 000, но так как большинство пациентов выживает, то количество заболевших достигает 1 на 10 000. Подвержены все возрастные группы.

Рис. 6. Нервномышечный синапс
Клинические проявления
- Птоз
- Диплопия с ограничением движения глазных яблок
- Слабость мимических мышц
- «Голос миастеника»
- Слабость при закрывании глаз
- Бульбарные нарушения:
- дисфагия (с попаданием пищи в носовые ходы)
- дизартрия (с носовым оттенком)
- Вовлечение дыхательных мышц (острые бульбарные и дыхательные расстройства, вызванные миастенией, требуют неотложной помощи)
- Слабость мышц шеи и конечностей, усиливающаяся к концу дня и после нагрузок («патологическая утомляемость»).
Обследование
- Определение содержания антител к ацетилхолиновым рецепторам в сыворотке крови (у 15% пациентов результат анализа отрицательный).
- Проба с введением антихолинэстеразных препаратов: преходящее и быстро нарастающее улучшение состояния после внутривенного введения эдрофониума (антихолинэстеразный препарат короткого действия, блокирует катаболизм ацетилхолина, временно повышая его содержание). (В Российской Федерации используется тест с прозерином). Тест более эффективен при использовании метода двойного контрольного исследования. Ввиду возможных холиномиметических эффектов вследствие повышения уровня ацетилхолина следует обеспечить возможность экстренного введения атропина и проведения реанимационных мероприятий.
- ЭМГ, включая игольчатое исследование с отведением потенциала от отдельных волокон.
- Исследование функции щитовидной железы при сопутствующем тиреотоксикозе.
- У пациентов с тимомой выявляются антитела в тканях поперечнополосатых мышц.
- КТ переднего средостения для выявления гиперплазии тимуса.
Лечение
- Антихолинэстеразные препараты, например пиридостигмин, в качестве симптоматического лечения. Пациентам требуется постоянное увеличение дозировок препаратов, что может привести к развитию холиномиметических побочных эффектов с повышением слюноотделения, рвотой, болью в эпигастрии и диареей. В редких случаях возможно развитие холинергического криза
- Кортикостероиды (преднизолон) назначаются при заболевании средней тяжести, не поддающемуся другому лечению. Лечение начинается с низких дозировок с постепенным увеличением дозы, препарат применяют через день. В начале лечения возможно нарастание симптоматики. Стационарное лечение показано в начале применения кортикостероидов у пациентов с генерализованными формами заболевания. По мере наступления эффекта доза может быть снижена в соответствии с клинической картиной.
- Иммуносупрессоры (азатиоприн) используются в комбинации с кортикостероидами при заболевании средней тяжести.
- Тимэктомия показана при тимоме и у молодых пациентов на ранних стадиях заболевания для уменьшения потребности в лекарственной терапии и реже — для достижения полной ремиссии.
- Плазмаферез или внутривенное введение иммуноглобулина как средства подготовки к тимэктомии и при тяжелых формах заболевания.
Пациентам с миастенией следует избегать приема некоторых антибиотиков, таких как аминогликозиды, вследствие их блокирующего эффекта на уровне нейромышечного синапса.
Другие миастенические синдромы
Реже нейромышечный синапс может страдать в результате наследственного заболевания или вследствие наранеопластического процесса (миастенический синдром Ламберта-Итона).
Миопатии
Основные причины развития миопатий приведены в табл. 5. Клинически миопатия проявляется слабостью мышц туловища и проксимальных отделов конечностей. Возможны слабость мимических мышц и мускулатуры шеи, выявляемая при сгибании и/или разгибании. Походка становится неустойчивой. При приобретенном характере заболевания мышечная слабость может быть относительно умеренной, по крайней мере на ранних стадиях, а сухожильные рефлексы на протяжении длительного времени остаются сохранными.
Таблица 5. Причины возникновения миопатий
|
Наследственные факторы Мышечная дистрофия Метаболические миопатий Инфекционные заболевания Газовая гангрена Стафилококковый миозит Вирусная инфекция (вирусы гриппа, Коксаки, ЕСНО) Паразиты (цистицеркоз, трихинеллез) Воспалительные процессы Полимиозит Дерматомиозит Саркоидоз Новообразования Дерматомиозит - может быть следствием паранеопластического процесса Метаболические (приобретенные) расстройства Тиреотоксикоз Синдром Кушинга Остеомаляция Токсикоз (от приема лекарственных препаратов) Кортикостероиды Галотан - злокачественная гипертермия (редко) Другие лекарственные препараты |
Обследование больного с миопатией:
- анализ крови:
- СОЭ, аутоантитела (при приобретенных заболеваниях)
- креатинкиназа — уровень резко повышен вследствие высвобождения из поврежденных мышечных клеток
- ЭМГ
- биопсия мышц.
Клинические синдромы
Мышечные дистрофии
Дистрофинопатии
Заболевание обусловлено мутацией гена, связанного с Х-хромосомой и отвечающего за синтез мышечного белка дистрофина. Встречается у детей, подростков и у взрослых. Детская форма (мышечная дистрофия Дюшена) протекает наиболее тяжело. У заболевших мальчиков уже в раннем детстве развивается слабость в проксимальных отделах конечностей. Характерным признаком является взбирание «лесенкой» (симптом Говерса). Мышцы голеней могут казаться гипертрофированными (рис. 7) из-за замещения мышечных волокон соединительной тканью (псевдогипертрофия). Дети обычно прикованы к креслу-каталке до подросткового возраста. Болезнь неуклонно прогрессирует, смерть наступает от сердечных или дыхательных осложнений в возрасте до 20 лет. Менее тяжелое течение наблюдается при дебюте заболевания в подростковом или взрослом возрасте (мышечная дистрофия Беккера). Заболевание, как правило, не носит угрожающего для жизни характера, однако зачастую сопряжено с прогрессирующей инвалидизацией. В настоящее время имеется возможность диагностики миодистрофий при помощи молекулярного анализа гена дистрофина.

Рис. 7. Псевдогипертрофия голеней
Другие мышечные дистрофии
Миотоническая дистрофия — заболевание с аутосомно-доминант-ным типом наследования, при котором у пациентов присутствует аномально длительное напряжение мышц (миотония). Проявляется невозможностью расслабления мышцы. Характерным признаком является перкуссионная миотония, которая выявляется постукиванием молоточком по мышце. Миотония может быть диагностирована при электромиографическом обследовании.
Типичные симптомы:
- двусторонний птоз
- слабость лицевых мышц
- паралич и слабость грудино-ключично-сосцевидных мышц
- ранняя катаракта
- сопутствующие эндокринные нарушения (сахарный диабет, облысение и атрофия яичек).
В качестве терапии при миотонии могут применяться фенитоин или мексилетин. При наследственной миотонии наблюдаются умеренно выраженные атрофия и слабость мышц.
Лице-лопаточно-плечевая мышечная дистрофия является аутосомно-доминантным заболеванием. У пациентов наблюдается двусторонняя слабость мимических мышц и крыловидное расположение лопаток. В дополнение к параличу и слабости проксимальных мышц верхних конечностей обычно имеются слабость мышц спины и тазового пояса, наблюдается неустойчивая походка и поясничный гиперлордоз. Реже при мышечных дистрофиях и врожденных миопатиях поражаются мышцы глазного яблока и глотки.
Другие наследственные заболевания
Метаболические нарушения, например гликогенозы, могут сопровождаться мышечной слабостью, нередко ассоциированной с миалгиями и спазмами.
При семейном периодическом параличе приступы выраженной мышечной слабости могут быть спровоцированы напряжением, употреблением пищи с высоким содержанием углеводов, длительным пребыванием на холоде. Заболевание может быть связано с гипо- и гиперкалиемией.
Приобретенные заболевания
Воспалительные миопатии
Полимиозит может развиваться как изолированно, гак и вместе с другими аутоиммунными поражениями соединительной ткани, например системным склерозом, фиброзирующим альвеолитом и синдромом ПТегрена.
Дерматомиозит является сопутствующим заболеванием при воспалительной миопатии с характерной фиолетовой (гелиотропной) сыпью на лице. Ярко-красная сыпь может быть локализована в области суставов, передней поверхности грудной клетки, поверхностях разгибателей. У некоторых пациентов с дерматомиозитом, в частности у мужчин старше 45 лет, нередко имеется злокачественное новообразование, например карцинома бронхов или желудка. -
Клинические проявления воспалительной миопатии такие же, как при проксимальной миопатии, однако могут также присутствовать дисфагия как результат вовлечения мышц глотки, мышечные боли и гиперестезия. Возможны также артралгия и феномен Рейно.
После гистологического подтверждения диагноза проводится лечение кортикостероидами и иммуносупрессорами (например, азатиоприн). Пациентам требуется наблюдение в течение нескольких лет, у многих остается мышечная слабость. Один из гистологически диагностируемых вариантов заболевания — миозит с включением телец — лечению не поддается. Данное состояние — довольно частая форма приобретенных мышечных заболеваний, поражающая в основном пожилых людей. Характерной особенностью является избирательное поражение сгибателей пальцев кистей и четырехглавых мышц. Недостаточный эффект применения иммуносупрессоров послужил основой гипотезы о вторичности воспалительных реакций по отношению к дегенеративным изменениям мышечной ткани.
Неврология для врачей общей практики. Л. Гинсберг
medbe.ru
Издания
Лекции и обзоры
С.В. РевенкоГармонические перспективы реографии В обзоре кратко описаны основные пути развития реографии. Особое внимание уделено практически забытому и вновь возрожденному методу гармонического анализа реограмм. Развитие электроники и компьютерных методов обработки данных позволило раскрыть потенциал этого направления реографии и рассмотреть перспективы разработки новых диагностических подходов на основе мультицикличного гармонического анализа биоимпеданса.
М.Л. НовиковТравматические повреждения плечевого сплетения: современные способы хирургической коррекции.Часть I. Диагностика повреждений плечевого сплетения Задача настоящей публикации – познакомить практикующих неврологов, нейрохирургов, травматологов и ортопедов с современными принципами диагностики и лечения различных повреждений плечевого сплетения (ППС). Подробно описана анатомия плечевого сплетения, рассматриваются основные механизмы его повреждения, дается их современная классификация. Особое внимание уделено тракционному механизму ППС, как наиболее сложному и часто встречающемуся и основному в группе пациентов, нуждающихся в оперативном лечении. Рассматриваются возможности различных инструментальных методов – рентгенологических, нейровизуализационных, электрофизиологических – в диагностике ППС. Предлагается авторский алгоритм дифференциально-диагностического поиска при данной патологии.
Fausto Machicao, Dafin Fior Muresanu, Harald Hundsberger, Maren Pfluger, Alla GuekhtПлейотропный нейропротективный и метаболический эффекты актовегина В обзоре рассматриваются механизмы действия актовегина в контексте изучения его эффектов на доклиническом уровне и новой концепции фармакологического лечения неврологических расстройств. Актовегин, получаемый при ультрафильтрации крови телят, состоит из более чем 200 биологических субстанций. Препарат используется при широком спектре заболеваний, включая нарушения периферического и мозгового кровообращения, ожоги, плохое заживление ран, радиационные поражения и диабетическую полинейропатию. Актовегин состоит из молекул малого размера, которые находятся в организме в нормальных физиологических условиях, и поэтому исследования их фармакокинетики и фармакодинамики для определения активной субстанции препарата затруднены. Результаты преклинических исследований показали, что актовегин улучшает метаболический баланс путем повышения усвоения глюкозы и потребление кислорода в условиях ишемии. Актовегин также повышает устойчивость к гамма-радиации и стимулирует ранозаживление. В более поздних работах было установлено, что антиоксидантный и антиапоптотический механизмы действия лежат в основе нейропротективных свойств актовегина, подтвержденных в экспериментах на первичных нейронах гиппокампа крыс и стрептозотоцининдуцированной модели диабетической полинейропатии у крыс. Последние данные свидетельствуют о положительном влиянии актовегина на фактор NF-?B, но при этом многие молекулярные и клеточные механизмы его действия остаются неизвестными. В первую очередь это касается влияния актовегина на нейропластичность, нейрогенез и трофическую функцию нервной системы, и данный аспект требует дальнейших исследований. Тем не менее становится очевидным, что мультифакториальная и многокомпонентная природа актовегина определяет его плейотропный нейропротективный механизм действия и клиническую эффективность.
Оригинальные исследования.
Н.Г. Савицкая, А.В. Остафийчук, Н.А. Супонева, Д.С. ЯнкевичВозможности электромиографии в прогнозировании восстановления при идиопатической нейропатии лицевого нерва Представлены результаты ретроспективного клинико-электромиографического (ЭНМГ) анализа 182 пациентов с идиопатической нейропатией лицевого нерва (НЛН). Проведено сравнение наиболее часто исследуемых ЭНМГ-параметров для определения благоприятного и неблагоприятного прогнозов восстановления. Выявлено, что наиболее чувствительным ЭНМГ-параметром в острейшем периоде (до 5 дней) НЛН является определение порога вызывания моторного ответа и исследование мигательного рефлекса; в остром периоде (от 10 до 14 дней) – измерение процентного соотношения падения амплитуды М-ответа пораженной стороны по отношению к здоровой; начиная с 21 дня – наличие неврогенных изменений в мышцах. На основании полученных данных предлагается наиболее оптимальный объем ЭНМГ-исследования при идиопатической НЛН на разных сроках заболевания.
А.Л. Куренков, А.Р. АртеменкоРоль периферического нейромоторного аппарата в формировании тяжелых двигательных нарушений у больных с детским церебральным параличом Двигательный стереотип у пациентов с детским церебральным параличом (ДЦП) определяется выраженностью спастичности и центральных парезов мышц, нарушениями механизмов межмышечного взаимодействия, а также наличием содружественных реакций и патологических синкинезий. Имеются наблюдения сочетания при ДЦП поражения надсегментарных и сегментарных структур. Предпринята попытка выявления поражения периферического нейромоторного аппарата и вклада обнаруженных нарушений в клиническую картину ДЦП в поздней резидуальной стадии. При ДЦП в форме спастической диплегии с прогрессирующими деформациями суставов нижних конечностей в 12,3 % обнаруживается поражение центральной нервной системы на центральном и сегментарном уровнях. Игольчатая электромиография – наиболее чувствительный метод выявления поражения на сегментарном уровне: регистрируются потенциалы двигательных единиц (ПДЕ) увеличенной длительности и амплитуды, уменьшение числа рекрутируемых ПДЕ. При турн-амплитудном анализе облачная диаграмма смещается влево и вверх, уменьшается отношение числа турнов к средней амплитуде турнов. Резидуальный характер изменений подтверждается отсутствием признаков текущего денервационного процесса. Обсуждаются вклад миелодисплазии и транснейрональной дегенерации спинальных мотонейронов пояснично-крестцового утолщения спинного мозга в клинику рассматриваемой формы ДЦП и тактика реабилитационных и ортопедо-хирургических мероприятий.
Клинический разбор
А.Г. Санадзе, Л.Ф. КасаткинаДва случая трансформации миастении в боковой амиотрофический склероз Описаны 2 случая трансформации миастении в боковой амиотрофический склероз (БАС). У 72-летней женщины и 38-летнего мужчины выявлены достоверные клинические признаки генерализованной миастении, с наличием глазодвигательных расстройств и птоза, нарушением бульбарных функций, слабостью мышц лица и конечностей. Диагноз миастении подтверждался результатами фармакологического теста, электромиографического (ЭМГ) тестирования нервно-мышечной передачи, повышением концентрации аутоантител к ацетилхолиновому рецептору, титин-протеину, а также эффектом глюкокортикоидной терапии и тимэктомии. У обоих пациентов в интервале от 4 до 7 лет от начала болезни появились ЭМГ- и клинические симптомы БАС.
С.А. Мальмберг, Е.Н. РуденкоЛюмбосакральная моторная полиневропатия Представлено описание случая люмбосакральной моторной полиневропатии (ЛСМП) у пациентки 15 лет, страдающей сахарным диабетом 1-го типа. Подробно рассмотрены клиническая картина и электронейромиографические изменения при данной форме болезни, оценена эффективность проведенной кортикостероидной терапии. Обсуждены вопросы дифференциального диагноза и таксономической позиции, сближающей ЛСМП с хронической воспалительной демиелинизирующей полиневропатией (ХВДП). Рассматривается необходимость некоторой либерализации диагностических критериев ХВДП.
Н.П. Котлукова, С.В. Михайлова, Т.М. Букина, Е.Ю. Захарова.Младенческая форма болезни Помпе: клиника, диагностика и лечение Болезнь Помпе является редким (орфанным) наследственным заболеванием, которое относится к лизосомным болезням накопления и может рассматриваться как сердечный гликогеноз II типа, а также как тяжелое нервно-мышечное заболевание или метаболическая миопатия. Выявляемость данной патологии среди врачей различного профиля крайне низкая, что обусловлено как редкостью патологии, так и клинико-генетическим полиморфизмом заболевания. Наиболее тяжелая форма болезни Помпе - младенческая (инфантильная). Она характеризуется прогредиентностью течения и летальным исходом в течение первого года жизни. Возможность проведения фермент-заместительной терапии при данном заболевании, позволяющая улучшить прогноз и качество жизни пациентов, определяет актуальность ранней диагностики болезни Помпе. В статье описываются клиника, современные методы диагностики и лечения инфантильной формы болезни Помпе. Представлен собственный опыт диагностики и лечения младенческой формы болезни Помпе на основании демонстрации 3 клинических случаев заболевания. Обсуждаются особенности каждого ребенка, подтверждающие клинико-генетическое разнообразие данной патологии.
Конференции, симпозиумы, совещания
Отчет о симпозиуме "Общества специалистов по нервно-мышечным болезням" в рамках конференции НП "Объединение врачей-эпилептологов и пациентов". Конференция "Инновации в эпилептологии III+" проходила 8–10 ноября в Санкт-Петербурге и Москве в два этапа. Особенность конференции заключалась в участии в ней и врачей, и пациентов.
Выдающиеся российские неврологи
Л.А. СайковаВладимир Семенович Лобзин. Жизненный путь и творческая деятельность. История отечественной неврологии и в частности заболеваний периферических нервов и мышц тесно связана с именем выдающегося невролога Владимира Семеновича Лобзина (1924–1993). В.С. Лобзин внес весомый вклад в разработку проблемы нервно-мышечных заболеваний и создал целую научную школу миологии. За скромными сведениями приводимой ниже биографии и перечислением основных трудов стоит жизнь талантливого человека. Обладавший необычайной работоспособностью и ярким дарованием ученого-исследователя, Владимир Семенович оставил большое научное наследие. Его исследования успешно продолжаются учениками и коллегами. В 1982–1992 гг. В.С. Лобзин возглавлял кафедру невропатологии Санкт-Петербургской медицинской академии последипломного образования, которой позже было присвоено имя академика АМН СССР С.Н. Давиденкова, чему способствовал при жизни Владимир Семенович.
abvpress.ru
Наши издания
Введение. Краткий обзор состояния вопроса со ссылками на наиболее значимые публикации, причина необходимости проведения исследования.
Цели. 2-3 предложения о том, какую проблему или гипотезу решает автор и с какой целью.
Материалы и методы. Подробное изложение методик исследования, аппаратуры, критериев отбора участников, их число и характеристики, способы и принципы распределения на группы, дизайн исследования, методы статистического анализа. Описанные методы исследования должны гарантировать возможность воспроизведения результатов. Величины измерений должны соответствовать Международной системе единиц (СИ). При перечислении использованной аппаратуры и препаратов в скобках указываются производитель и страна; при перечислении используемых в ходе работы лекарственных препаратов и химических веществ - их международное непатентованное (общепринятое) название, дозы, пути введения.
Если того требует проведенное исследование, указать решением какого этического комитета оно одобрено; а также факт подписания испытуемыми информированного согласия.
Результаты. Должны быть представлены в логической последовательности, отражать данные описанного выше исследования без ссылок на литературные источники. Результаты представляются чётко, в виде коротких описаний с указаниями на графики, таблицы и рисунки.
Обсуждение. Выделение новых и важных аспектов по результатам проведенного исследования, анализ возможных механизмов или толкование полученных результатов. По возможности сопоставление собственных результатов с данными других исследователей. Возможно включение обоснованных рекомендаций для клинической практики и применения полученных данных в предстоящих исследованиях.
Следует избегать повторения сведений из раздела «Введение» и подробного перечисления данных из раздела «Результаты».
Заключение. Должно быть кратким и лаконичным (не более одного абзаца). Подведение итога проделанной работы и гипотеза авторов о значении полученных данных - в рамках патогенеза, лечения, диагностики; перспективы использования полученных данных.
7. Иллюстративный материал.
Иллюстративным материалом являются фотографии, рисунки, схемы, графики, диаграммы, таблицы.
Иллюстративный материал должен быть представлен в виде отдельных файлов и не фигурировать в тексте статьи (схемы, графики, диаграммы, таблицы могут быть собраны в общие файлы по типу иллюстративного материала с началом каждого с новой страницы.
Например: все таблицы собраны в отдельный от статьи файл, где каждая новая таблица начинается с новой страницы. Если в этой же статье есть диаграммы, то они будут составлять следующий файл, где будут собраны исключительно диаграммы, и каждая из них будет размещена на отдельном листе документа). Файлы иллюстративного материала должны позволять воспроизвести высокое качество изображения в электронной и печатной версии журнала. Если иллюстративный материал ранее был опубликован в других изданиях, автор обязан предоставить в редакцию разрешение правообладателя на публикацию данного изображения в другом журнале, в противном случае это будет считаться плагиатом и к публикации принято не будет. Количество иллюстраций должно соответствовать объему предоставляемой информации, избыточность иллюстраций может привести к возвращению авторам статьи для доработки на предмет сокращения. Данные таблиц не должны повторять данные рисунков и текста и наоборот.
Фотографии представляются в формате TIFF, JPG, CMYK с разрешение не менее 300 dpi (точек на дюйм). Разрез глаз пациентов или здоровых испытуемых на фотографиях должны быть закрыты чёрным прямоугольником, в случае его отсутствия автор должен предоставить в редакцию письменное разрешение пациента на публикацию.
Рисунки, графики, схемы, диаграммы представляются в формате EPS Adobe Illustrator 7.0— 10.0 или Office Excel. При невозможности представления в данном формате необходимо связаться с редакцией.
Все рисунки должны быть пронумерованы и снабжены подрисуночными подписями. Фрагменты рисунка обозначаются строчными буквами русского алфавита — «а», «б» и т.д. Все сокращения, обозначения в виде кривых, букв, цифр и т.д., использованные на рисунке, должны быть расшифрованы в подрисуночной подписи. Подписи к рисункам даются на отдельном листе после текста статьи в одном с ней файле.
Таблицы должны быть наглядными, иметь название и порядковый номер. Заголовки граф должны соответствовать их содержанию. Все сокращения расшифровываются в примечании к таблице. Необходимо указывать применявшийся для анализа статистический метод и соответствующее значение достоверности (р). В случае размера таблиц больше, чем на лист А4, они представляются в виде отдельного файла doc, docx, rtf.
8. Единицы измерения и сокращения.
Единицы измерения даются в Международной системе единиц (СИ). Если исследование проводилось на приборах, дающих показатели в других единицах, необходимо перевести их в систему СИ с указанием коэффициента пересчета или компьютерной программы в разделе «Материалы и методы».
Сокращения слов не допускаются, кроме общепринятых. Все аббревиатуры в тексте статьи должны быть полностью расшифрованы при первом упоминании (например, нервно-мышечные болезни (НМБ)).
Название генов пишется курсивом, название белков — обычным шрифтом.
9. Список литературы.
На следующей странице после текста статьи должен располагаться список цитируемой литературы.
Литература приводится в порядке цитирования. Все источники должны быть пронумерованы, нумерация осуществляется строго по мере цитирования в тексте статьи, но не в алфавитном порядке. Все ссылки на литературные источники в тексте статьи печатаются арабскими цифрами в квадратных скобках (например [5]). Количество цитируемых работ: в оригинальных статьях желательно не более 20–25 источников, в обзорах литературы — не более 60.
Ссылка должны даваться на первоисточники и не цитировать один обзор, где они упомянуты. Ссылки на неопубликованные работы не допускаются.
Ссылки на литературные источники должны быть оформлены следующим образом:
Для каждого источника необходимо указать: фамилии и инициалы авторов (если авторов более 4, указываются первые 3 автора, затем ставится «и др.» в русском или «et al.» в английском тексте). Авторы цитируемых источников должны быть указаны в том же порядке, что и в первоисточнике.
Статья в журнале. Фамилия И.О. авторов. Название статьи. Название журнала год; том; номер выпуска: страницы (повторяющиеся цифры страниц не указывать, например: 185‒7). Обязательным для статей является указание индексов DOI и PMID (уникальный код статьи в PubMed) при их наличии. (Индекс DOI Вы можете узнать на сайте www.crossref.org).Примеры: Пирадов М.А., Супонева Н.А.. Аутоиммунные заболевания нервной системы: состояние проблемы и перспективы. Вестник РАМН 2015; 70 (2): 183–187. DOI: 10.15690/vramn.v70i2.1311 Wolf W.A., Martin J.L., Kartje G.L. et al.Evidence for fibroblast growth factor 2 as a mediator of amphetamine enhanced motor improvement following stroke. PLoS One 2014; 9 (9): e108031. DOI:10.1371/journal.pone.0108031. PMID:25229819
Монографии. Фамилия И.О. авторов. Полное название книги. Место издания: название издательства; год издания; номера страниц или общее количество страниц.Пример: Левин О.С. Полиневропатии. М.: МИА; 2015; 469 с. Fujimoto J.G., Brezinski M.E. Optical coherence tomography imaging. In: Biomedical photonics handbook. Vo-Dinh T. (editor). CRC Press; 2003; p. 22–24.
Ссылки на тезисы из сборника. Ссылки на тезисы возможны исключительно на зарубежные издания, опубликованные на английском языке. Фамилия И.О. авторов. Полное название тезиса. Название издательства. Название конференции. Место издательства; год издания; номера страниц.Пример: McMahan C., Hovland R., McMillan S. Gender and internet advertising: Differences in the ways males and females engage with and perceive internet advertising. In: S. Rodgers (Ed.), Proceedings of the 2008 Conference of the American Academy of Advertising. Columbia, MO: University of Missouri Press, 2008. pp. 52–55.
Ссылки на авторефераты диссертаций, неопубликованные работы, а также на данные, полученные из Internet, не допускаются.
10. Этические вопросы.
1) Авторство. Право называться автором имеют лица, которые: 1) внесли значительный вклад в концепцию и дизайн исследования или в анализ и интерпретацию данных; 2) активно участвовали в подготовке текста статьи или внесении принципиальных изменений; 3) участвовали в окончательном утверждении версии, которая сдается в печать; 4) готовы принять на себя ответственность за содержание статьи. Исключительно обеспечение финансирования или подбор материала для статьи не оправдывает включения в состав авторской группы. Общее руководство исследовательским коллективом также не признается достаточным для авторства. Редакторы вправе запросить информацию о вкладе каждого из авторов в написание статьи и опубликовать её. Все члены коллектива, не отвечающие критериям авторства, но оказавшие помощь в проведении исследования по сбору, анализу и интерпретации данных, предоставлению материалов и инструментов, должны быть перечислены с их согласия в разделе «Благодарности».
2) Конфликт интересов. В конце статьи необходимо указать наличие конфликта интересов для всех авторов. Конфликт интересов подразумевает наличие каких-либо связей и/или личной заинтересованности, которые потенциально могут повлиять на результаты, интерпретацию полученных данных, объективное их восприятие, в частности финансовые отношения и сотрудничество с какими-либо организациями (например, получение гонораров, образовательных грантов, участие в экспертных советах, членство, трудовые отношения, консультационная работа, владение магазином в частной собственности или другие интересы) или нефинансовая заинтересованность (например, личные или профессиональные взаимоотношения, знакомства и пр.), касающиеся рассматриваемых в статье вопросов и/или материалов. В случае отсутствия конфликта интересов в конце статьи следует констатировать следующее: «Авторы заявляют об отсутствии конфликта интересов».
3) Соблюдение прав больных и конфиденциальность. Больные имеют право на сохранение конфиденциальности, которую нельзя раскрывать без их согласия. Позволяющая установить личность информация, включая имена больных, инициалы, номера больниц и историй болезни, не должна публиковаться в виде письменных описаний, фотографий и родословных, если только эта информация не представляет большую научную ценность или если больной (или родитель, или опекун) не предоставит (предоставят) письменное согласие на публикацию. В таком случае авторы должны сообщить больным, существует ли вероятность того, что материал, позволяющий установить личность, после публикации будет доступен через Интернет. Авторы должны предоставить в редакцию письменное информированное согласие больного на распространение информации и сообщить об этом в статье.
4) Источник финансирования. В случае финансирования исследования, в конце статьи должен быть указан источник финансирования. При отсутствии такового, авторам стоит указать, что исследование проводилось без спонсорской поддержки.
11. Оплата публикации.
Все статьи, в том числе подготовленные аспирантами и соискателями ученой степени кандидата наук по результатам собственных исследований, принимаются к печати бесплатно.
Статьи, не соответствующие данным требованиям, к рассмотрению не принимаются.
Подготовка статей
Для представления статьи авторы должны проверить соответствие материала нижеследующим пунктам. В рассмотрении статьи редакцией авторам может быть отказано, если она им не соответствует.
www.neuromuscular.ru
Издания
Лекции и обзоры
Норма Беатриз Ромеро (Norma Beatriz Romero)Немалиновые миопатии: клиническое разнообразие и генетическая гетерогенность Врожденные миопатии составляют гетерогенную группу генетических мышечных патологий, вызванных структурной аномалией скелетной мышцы. Немалиновая миопатия принадлежит к обширной группе врожденных миопатий с белковыми включениями и характеризуется присутствием небольших включений в форме нитей, названных “rod”(англ., «стержень, прутик»). Речь идет о генетически обособленной группе, в которой идентифицированные основные ответственные гены кодируют белки тонких филаментов саркомеров. При этом сегодня гены определены лишь для 50 % известных случаев (ACTA1, NEB, TPM2, TPM3, TNNT1, CFL2 и KBTBD13). Последнее обстоятельство требует продолжения научных поисков в этой мало раскрытой области.
М.Л. Новиков, Т.Э. ТорноТравматические повреждения плечевого сплетения и современные способы хирургической коррекции. Часть II. Тактика лечения повреждений плечевого сплетения Задача настоящей публикации - познакомить практикующих неврологов, нейрохирургов, травматологов и ортопедов с современными принципами диагностики и лечения различных повреждений плечевого сплетения (ПС). В части I была подробно описана анатомия ПС, рассматривались основные механизмы его повреждения, была дана их совреме-нная классификация (Нервно-мышечные болезни 2012;4:19–27). В части II рассматриваются возможные варианты лечения пациентов на всех этапах оказания медицинской помощи: определение показаний для консервативного или хирургического лечения, предоперационное ведение, реабилитационное лечение. Подробно раз бираются тактика и техника первичных хирургических реконструкций.
Н.А. СупоневаКлиническая и диагностическая роль аутоантител к ганглиозидам периферических нервов: обзор литературы и собственные данные Исследование антител к гликолипидам периферических нервов стало доступно для широкой практики во многих городах России. Показанием к проведению диагностического теста на определение антиганглиозидных антител является подозрение на синдром Гийена-Барре, синдром Миллера Фишера, энцефалит Бикерстаффа, хроническую воспалительную демиелинизирующую полинейропатию, мультифокальную моторную нейропатию. Показанием к определению анти-MAG-антител служит наличие у больного IgM- парапротеинемической полинейропатии. Зарубежный и отечественный опыт подтверждает необходимость включения этих иммунологических тестов
Оригинальные исследования.
А.В. Сахарова, Л.В. Диденко, Т.И. Муравина, Р.П. Чайковская, Е.А. Кост, М.Ф. Мир-КасимовПрямая морфологическая детекция Borrelia burgdorferi в мышечных биоптатах: возможность связи нервно-мышечной патологии с боррелиозом Исследовано 40 биоптатов мышц, взятых у больных с нервно-мышечной симптоматикой при неустановленном или предположительном диагнозе. В 18 из них в полутонких срезах ткани обнаружены очаги повреждения мышечных волокон с наличием спирохетоподобных структур. При электронной микроскопии этих участков были выявлены боррелии в виде вегетативных и разнообразных L-форм. Применение методов иммуноцитохимии с антителами к антигенам Borrelia burgdorferi подтвердило принадлежность спирохет к данному виду. Это позволило считать этиологическим или осложняющим фактором нервно-мышечной патологии боррелиоз, а также рекомендовать применять указанные морфологические методы как диагностические в случаях нервно-мышечных заболеваний неясного происхождения.
Клинический разбор
Н.А. Шнайдер, Т.Я. Николаева, Е.Н. Бороева, Г.М. Пшенникова, Н.В. Лугинов, Ю.С ПанинаКонечностно-поясная мышечная дистрофия с аутосомно-доминантным типом наследования: пельвиофеморальная форма Лейдена–Мебиуса В статье рассматриваются современные подходы к клинико-лабораторной диагностике конечностно-поясной мышечной дистрофии с акцентом на аутосомно-доминантные формы заболевания. Авторами представлено собственное клиническое наблюдение случая с поздней диагностикой пельвиофеморальной формы конечностно-поясной мышечной дистрофии с аутосомно-доминантным типом наследования у пациентки 37 лет.
Выдающиеся российские неврологи
Д.И. Руденко, В.М. Казаков, Т.Р. СтучевскаяВладимир Карлович Рот (1848–1916) Владимир Карлович Рот является основоположником учения о нервно-мышечных болезнях в России. Он был одним из лучших учеников А.Я. Кожевникова, положившего начало отечественной неврологии. В.К. Рот, великий клиницист и патологоанатом, был блестящим знатоком электродиагностики и электротерапии нервно-мышечных болезней.
Конференции, симпозиумы, совещания
Отчет о проведении I Учредительной конференции регионального «Общества специалистов по нервно-мышечным болезням» 22–23 ноября 2012 г. в Москве состоялась I Учредительная конференция региональной общественной организации «Общество специалистов по нервно-мышечным болезням» (РОО ОНМБ), посвященная актуальным вопросам диагностики и лечения нервно-мышечных заболеваний в России. Мероприятие проводилось на базе Федерального научно-клинического центра специализированных видов медицинской помощи и медицинских технологий ФМБА России. На конференции присутствовало более 350 делегатов из Москвы, Самары, Ярославля, Иваново, Нижнего Новгорода, Красноярска, Санкт-Петербурга, Казани, Воронежа и других российских городов , а также из ближнего зарубежья – Украины, Белоруссии, Казахстана. Уникальность мероприятия заключалась в том, что впервые за последние десятилетия на одной площадке встретились все ведущие российские клиницисты в области заболеваний периферического нейромоторного аппарата, клинические нейрофизиологи, генетики и специалисты по нейровизуализации периферических нервов и мышц.
Отчет о конференции «Порфирия: особенности клиники, диагностики и лечения» 7 декабря 2012 г. в Москве состоялась конференция, посвященная одному из тяжелейших критических состояний в неврологии – порфирийной полинейропатии, ее клиническим особенностям, диагностике и современным принципам лечения. Организаторами этого мероприятия выступили ФГБУ «Научный центр неврологии» (НЦН) РАМН, Международная ассоциация организаций в области неврологии и нейронаук, ФГБУ «Гематологический научный центр» (ГНЦ) Минздрава России.
Англо-русский толковый словарь наиболее употребительных нейрофизиологических терминов. Часть I: A–D
abvpress.ru









